

Pacienții români cu fibroză chistică vor avea acces la terapia cu modulator CFTR
Compania farmaceutică Vertex anunță rambursarea terapiei cu ORKAMBI (lumacaftor/ivacaftor) pentru 150 de pacienți eligibili cu fibroză chistică din România.
 Pacienții cu fibroză chistică, o boală genetică rară, care limitează speranța de viață, afectând plămânii, ficatul, tractul gastro-intestinal, pancreasul, sinusurile, glandele sudoripare și tractul reproductiv, beneficiază acum de rambursarea medicamentului ORKAMBI, transmite Vertex Pharmaceuticals, care anunță că a finalizat procesul de rambursare a tratamentului. Tratamentul va fi rambursat astfel pacienților care au două copii ale mutației F508del din gena CFTR – regulator de conductanță transmembranară a fibrozei chistice vor beneficia de rambursarea tratamentului cu modulatorul CFTR – ORKAMBI (lumacaftor/ivacaftor), terapie care, din 17 martie 2022, este accesibilă pacienților eligibili diagnosticați cu această boală, în România.
Pacienții cu fibroză chistică, o boală genetică rară, care limitează speranța de viață, afectând plămânii, ficatul, tractul gastro-intestinal, pancreasul, sinusurile, glandele sudoripare și tractul reproductiv, beneficiază acum de rambursarea medicamentului ORKAMBI, transmite Vertex Pharmaceuticals, care anunță că a finalizat procesul de rambursare a tratamentului. Tratamentul va fi rambursat astfel pacienților care au două copii ale mutației F508del din gena CFTR – regulator de conductanță transmembranară a fibrozei chistice vor beneficia de rambursarea tratamentului cu modulatorul CFTR – ORKAMBI (lumacaftor/ivacaftor), terapie care, din 17 martie 2022, este accesibilă pacienților eligibili diagnosticați cu această boală, în România.
„Este o etapă importantă pentru pacienții cu fibroză chistică din România și un alt pas în demersul Vertex de a oferi acces durabil la medicamentele noastre în întreaga lume. Suntem încântați că pacienții eligibili cu fibroză chistică din România au acum acces la ORKAMBI”, a declarat Amel Bech, Senior Country Manager Piețele de Distribuție Vertex, care a mulțumit „tuturor părților implicate în realizarea acestui demers, pentru colaborare, angajament și recunoașterea în mod susținut a valorii medicamentelor” produse de companie.
Fibroza chistică, o boală genetică gravă
 Fibroza chistică (CF), cunoscută și sub numele de mucoviscidoză, este o cea mai frecventă boală monogenică cu transmitere autosomal-recesivă, cauzată de anumite defecte ale genei CFTR ce codifică proteine ale canalelor de clor din membrana celulelor epiteliale și afectează peste 83.000 de oameni la nivel global.
Fibroza chistică (CF), cunoscută și sub numele de mucoviscidoză, este o cea mai frecventă boală monogenică cu transmitere autosomal-recesivă, cauzată de anumite defecte ale genei CFTR ce codifică proteine ale canalelor de clor din membrana celulelor epiteliale și afectează peste 83.000 de oameni la nivel global.
Prevalența bolii este de ~1,5 cazuri la 100.000 locuitori, iar frecvenţa purtătorilor de genă (heterozigoţi) este de 1 la 25. Diagnosticul de Fibroză chistică se pune de obicei în copilărie, frecvenţa bolii fiind estimată la 1/ 2500 de nou-născuţi. Copiii care moștenesc două gene CFTR defecte – una de la fiecare părinte – vor avea CF. În ultimii ani se raportează însă un numar din ce în ce mai mare de cazuri diagnosticate la vârsta adultă, iar mulţi pacienţi cu fibroză chistica ajung la vîrsta adultă şi necesită îngrijire de lungă durată.
Deși există mai multe tipuri de mutații CFTR care pot provoca boala, marea majoritate a persoanelor cu fibroză chistică au cel puțin o mutație F508del, care creează în fibroza chistică proteine CFTR nefuncționale și / sau prea puține la suprafața celulei. Acestea pot fi determinate printr-un test genetic sau test de genotipare. Funcția defectă și / sau absența proteinei CFTR cauzează un flux slab de sare și apă în și din celule în mai multe organe. În plămâni, acest lucru duce la acumularea de mucus anormal de gros, lipicios, care poate provoca infecții pulmonare cronice și leziuni pulmonare progresive la mulți pacienți, putând cauza moartea acestora. Vârsta medie a persoanelor decedate este de 30 de ani.
Fibroza chistică în România
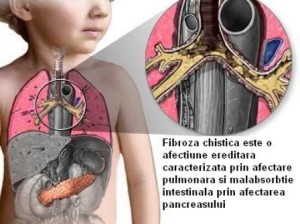
 În România, aproape 500 de pacienți au fost diagnosticați cu mucoviscidoză, o boală progresivă, multi-sistemică, ce afectează plămânii, ficatul, tractul gastro-intestinal, sinusurile, glandele sudoripare, pancreasul și tractul reproductiv. Toate aceste cazuri au fost confirmate prin teste genetice, dar întrucât nu există un program de screening neonatal, este dificil de spus care este incidența FC în România. Este posibil ca Registrul Național de FC să nu includă toți pacienții cu FC care trăiesc în România, deși se preconizează că numărul pacienților cu mutații de sincronizare să fie foarte scăzut.
În România, aproape 500 de pacienți au fost diagnosticați cu mucoviscidoză, o boală progresivă, multi-sistemică, ce afectează plămânii, ficatul, tractul gastro-intestinal, sinusurile, glandele sudoripare, pancreasul și tractul reproductiv. Toate aceste cazuri au fost confirmate prin teste genetice, dar întrucât nu există un program de screening neonatal, este dificil de spus care este incidența FC în România. Este posibil ca Registrul Național de FC să nu includă toți pacienții cu FC care trăiesc în România, deși se preconizează că numărul pacienților cu mutații de sincronizare să fie foarte scăzut.
În ultimele nouă săptămâni, pacienții cu fibroză chistică (mucoviscidoză) au protestat în fața Ministerului Sănătății, cerând să li se ramburseze noile terapii indicate în tratarea mucoviscidozei (fibrozei chistice), cu status homozigot al mutației F508del la nivelul genei responsabile de producerea proteinei cunoscute sub denumirea de reglator al conducție transmembranare în fibroza chistică (cystic fibrosis transmembrane conductance regulator = CFTR) sau cu status heterozigot al mutației dF508del la nivelul genei CFTR cu mutație de tip MF (reducere la minim a funcției).
Vineri, producătorul a anunțat că a finalizat procesul de rambursare a tratamentului pentru ORKAMBI, iar 150 de pacienți vor avea, de acum încolo, acces la tratament, această terapie, ca și alte terapii combinate, nefiind recomandate decât după vârsta de 12 ani, potrivit specificațiilor ANMDM.
Terapiile combinate pentru Fibroza chistică
 Pentru persoanele cu două copii ale mutației F508del, proteina CFTR nu este procesată în mod corespunzător în interiorul celulei, lucru care cauzează prezența acestei proteine într-un procent foarte mic sau chiar absența ei la suprafața celulei. ORKAMBI (lumacaftor / ivacaftor) este o terapie combinată între lumacaftor și ivacaftor.
Pentru persoanele cu două copii ale mutației F508del, proteina CFTR nu este procesată în mod corespunzător în interiorul celulei, lucru care cauzează prezența acestei proteine într-un procent foarte mic sau chiar absența ei la suprafața celulei. ORKAMBI (lumacaftor / ivacaftor) este o terapie combinată între lumacaftor și ivacaftor.
Lumacaftor, un modulator CFTR cunoscut drept corector, este creat pentru a crește cantitatea de proteină matură la suprafața celulei prin detectarea defectului de procesare și trafic al proteinei F508del-CFTR.
Ivacaftor, cunoscut ca un potențiator CFTR, este conceput pentru a spori funcția proteinei CFTR de a transporta sare și apă odată ce ajunge la suprafața celulei. Acțiunile combinate ale corectorului CFTR și ale potențiatorului ajută la hidratarea și eliminarea mucusului de la nivelul căilor respiratorii.
* Compania Vertex are mai multe medicamente aprobate care tratează cauza principală a fibrozei chistice (CF), o boală genetică periculoasă, și are mai multe programe clinice și de cercetare în desfășurare pentru tratamentul fibrozei chistice. Medicamentele Vertex pentru fibroza chistică sunt rambursate în peste 20 de țări din lume, inclusiv Austria, Cehia, Danemarca, Franța, Germania, Republica Irlanda, Italia, Olanda, Spania, Suedia, Marea Britanie și SUA.












